
If I were a researcher and had to decide what to research, I would probably research myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). The disease is beyond fascinating (and scary) and I personally know quite a few people who have had their lives derailed by it.
ME/CFS is probably the most impactful disease most people have never heard of. It affects a little above 1% of the population and it destroys the life of pretty much everyone who has it.
Last week, I was discussing medical topics with a friend and, as it happens so often, one potential idea of what ultimately causes the disease came from the dialectic back and forth of a simple conversation. The answer to what causes ME/CFS (a condition with a plethora of disparate symptoms), which had been puzzling researchers for years, might actually come from a very common condition: obesity.
But first, let me give a quick background.
Background
Most people know ME/CFS by a different name – long COVID. I personally think that the two are identical and long COVID represents a subset of people with ME/CFS.
A good friend of mine, who is now in his third year of the disease, describes a profound and qualitatively distinct fatigue that is not relieved by rest. Even on relatively “good” days, energy availability is sharply limited. Activities that were once trivial (e.g., showering, preparing food, walking short distances, holding conversations, reading, or concentrating on a screen) may consume a significant fraction of available energy. He must carefully ration activity to avoid deterioration.
On most days, he has quite severe cognitive dysfunction, commonly referred to as “brain fog,” which is reported by the majority of patients. This includes slowed information processing, impaired attention, reduced working memory, word‑finding difficulties, and diminished executive function. In survey studies, over 80–90% of patients report cognitive impairment (or “brain fog”) as a core symptom.
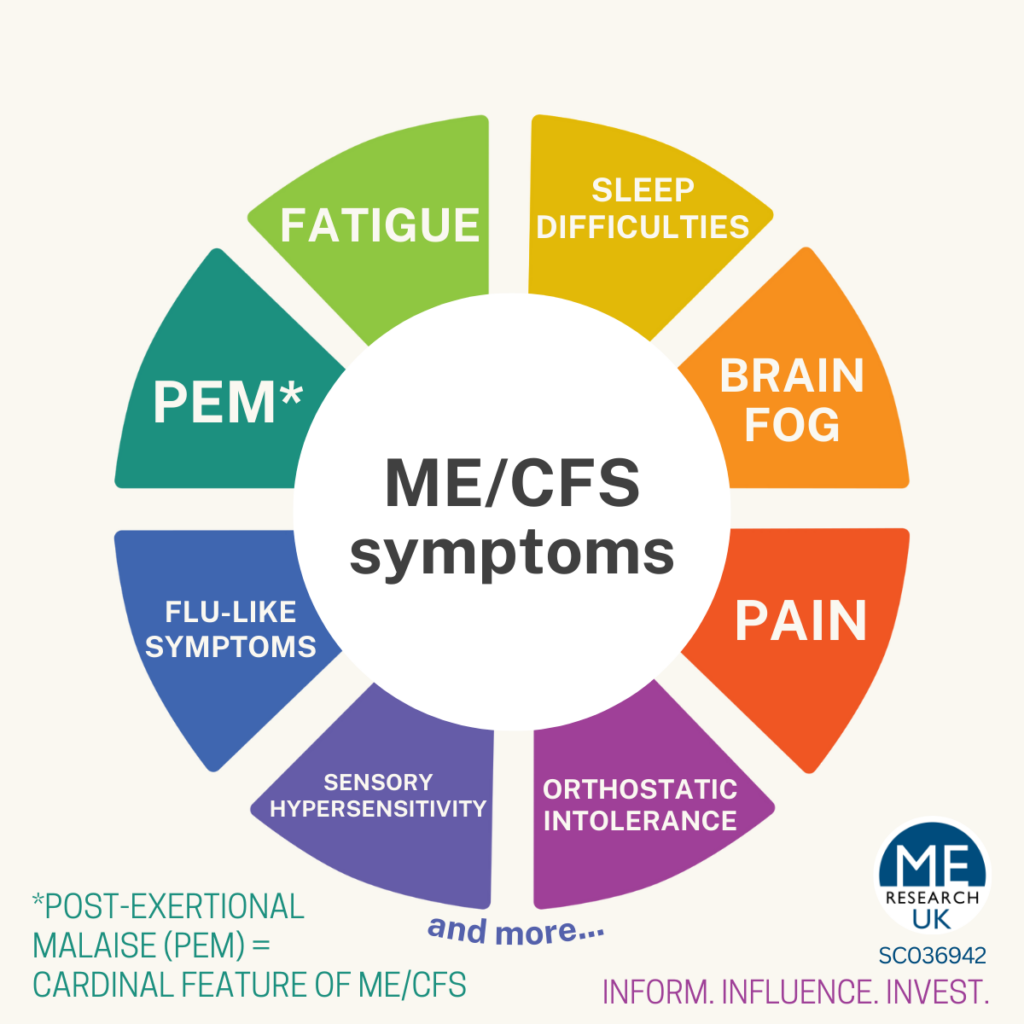
Sleep disturbances are nearly universal. He often sleeps 10–14 hours per day yet awakes unrefreshed. Polysomnographic studies reveal abnormalities in sleep architecture, including reduced slow‑wave sleep and frequent arousals.
Autonomic dysfunction is a central component of the illness. Between 50–80% of patients exhibit orthostatic intolerance, postural hypotension, or postural orthostatic tachycardia syndrome (POTS). Clinically, this manifests as dizziness, palpitations, presyncope, exercise intolerance, and exacerbation of symptoms when upright.
Neuropsychiatric symptoms are common but secondary. Anxiety, panic, depressive symptoms, depersonalization, and emotional lability frequently emerge after disease onset and correlate with severity.
For many patients, the lived experience of ME/CFS is one of persistent energy scarcity. Life becomes dominated by avoidance of crashes, uncertainty about recovery, and progressive restriction of activities.
Quality‑of‑life scores in severe ME/CFS are comparable to or worse than those observed in conditions such as multiple sclerosis, end‑stage renal disease, and congestive heart failure.
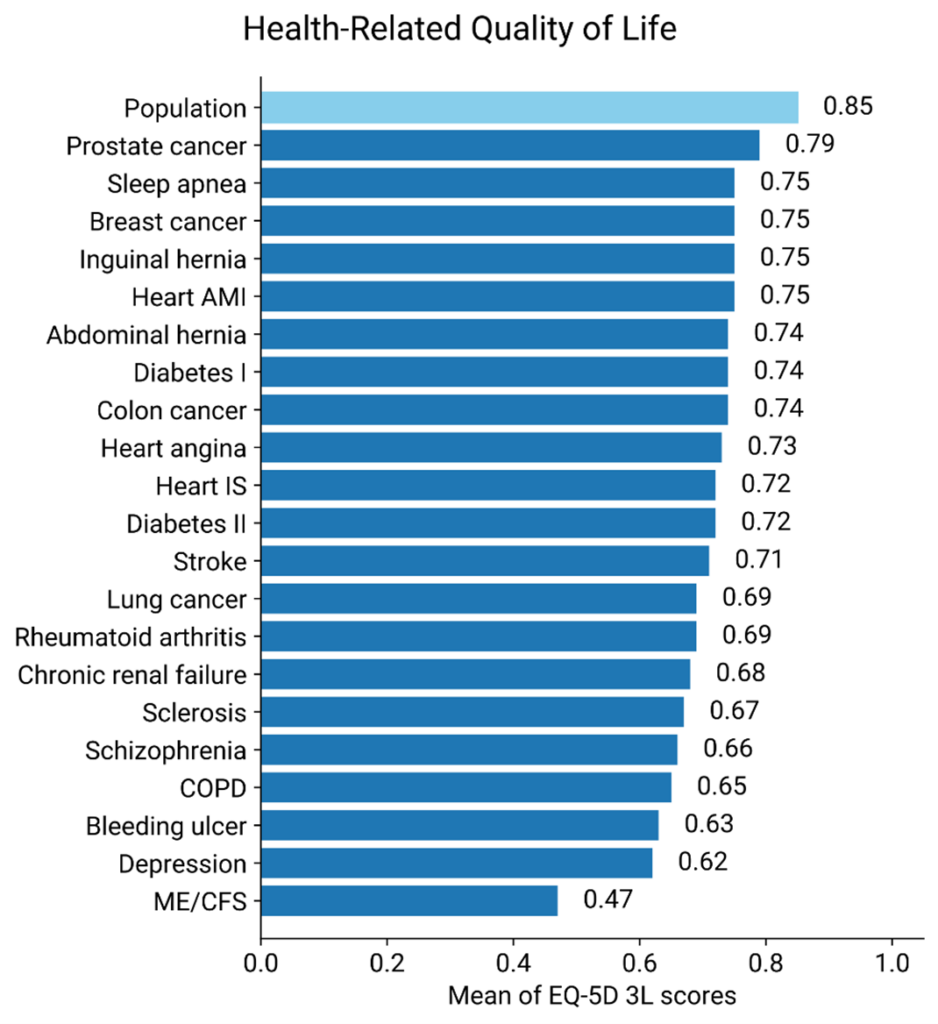
Globally, over 400 million people have experienced long-COVID, which is about 1 in 20. The prevalence of ME/CFS seems to be above 1%, which is huge. It is estimated that long COVID may be responsible for a loss of 1% of the world’s gross domestic product.
I personally know a number of individuals whose lives have been absolutely derailed for many months to years by Long-COVID. One of my best friends is two years after and he is still having waxing and waning periods with symptoms. Another acquaintance had to quit his high-paying consulting job, his girlfriend left him, and he is now vegetating in his apartment.
Another friend said that getting through Long-COVID without turning insane (his course lasted 3 years with many setbacks) is the biggest accomplishment in life. As I worked in the emergency apartment, I have also seen a couple of individuals whose life had been destroyed.
ME/CFS as a setpoint disease – The obesity connection
Everything that follows, is just a hypothesis. Nonethelss, one worth exploring because current ME/CFS research leads us nowhere.
To understand ME/CFS, we first need to understand obesity. Not in the way you might think (there is no direct link between the two conditions), but because obesity may be the best model we have for understanding what goes wrong in ME/CFS at the deepest level.
One of the most frustrating findings in obesity research is that the body defends its weight. When someone becomes obese and then loses weight through dieting or even through pharmacotherapy, the body fights to return to the higher weight. Leptin drops, the hypothalamus adapts, energy expenditure decreases, appetite increases, and these changes persist even after weight is restored to a normal range. The body has, in effect, adopted a new setpoint, a new “normal” that it will defend with remarkable tenacity.
This is why even patients on GLP-1 receptor agonists (drugs like semaglutide) tend to regain the weight once the medication is discontinued. The drugs work, but they work against the setpoint rather than resetting it. They are a workaround, not a cure.
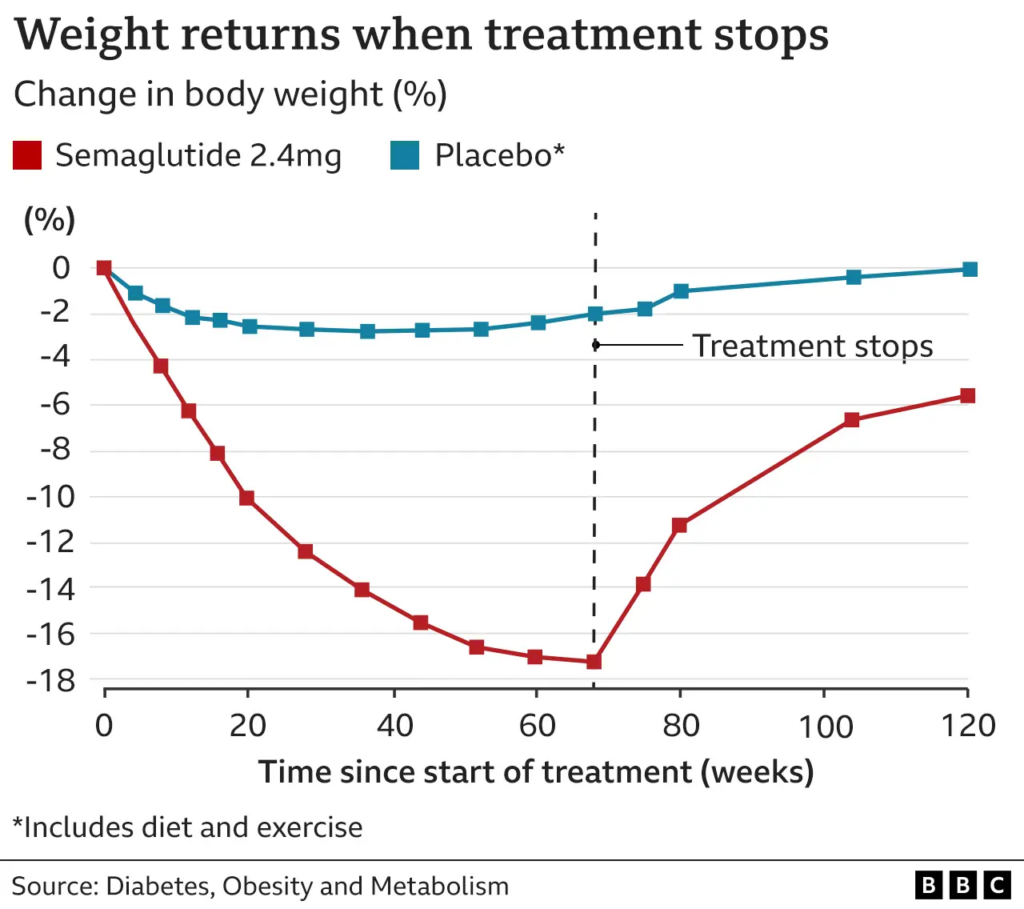
The mechanism behind this appears to be a two-layer system. At the deeper level, prolonged obesity causes epigenetic changes (alterations in how genes are read, without changing the DNA sequence itself) in key hypothalamic neurons. In particular, there is evidence of hypermethylation of the POMC promoter, which reduces the expression of POMC, a critical anorexigenic (appetite-suppressing) signal. On top of this altered epigenetic baseline, synaptic remodeling occurs; AgRP neurons (which drive hunger) gain more excitatory inputs, while POMC neurons gain more inhibitory inputs. I discuss POMC neurons in much more detail here: The Sickness of the Ultra-Fit
The synaptic remodeling is reversible on a timescale of days to weeks, but the underlying epigenetic changes are not. This is why short-term interventions (dieting, temporary pharmacotherapy) produce only temporary results: you are fighting against an epigenetically reprogrammed baseline that the synaptic architecture keeps returning to.
Mechanistically, the reprogramming presumably represents either DNA-hypermethylation of specific genes and/or self-sustaining loops of specific transcription factors, specifically turning up or down the expression level of a select set of genes associated with energy expenditure/body weight.
There is an even scarier possibility. The combination of hypothalamic inflammation and massive influx of metabolic signals (leptin, insulin, satiety peptides like GLP-1, CCK, PYY) may actually drive certain neurons, particularly POMC neurons, into excitotoxic states, leading to their death. If this is the case, obesity may partly reflect a state where a significant fraction of these critical neurons are simply gone.
GLP-1 agonists would then work by driving the remaining neurons into overdrive, compensating for reduced neuron numbers in much the same way that MAO-B inhibitors compensate for dopaminergic neuron loss in Parkinson’s disease. Fortunately, the balance of evidence suggests that functional impairment (epigenetic changes, leptin resistance, altered synaptic plasticity) is the dominant mechanism rather than outright cell death, which is good news because functional impairment is, at least in principle, reversible.
Though, as of yet, nobody was able to reverse obesity. However, obesity teaches us that setpoints can permanently change.
The core hypothesis – ME/CFS as a setpoint shift that fails to turn off
Now, here is the key insight that emerged from that conversation with my friend: what if ME/CFS represents exactly the same kind of phenomenon, a maladaptive setpoint shift, but in a different system?
The clinical pattern fits remarkably well. ME/CFS typically begins with an acute trigger (a viral infection like EBV or COVID, physical trauma, severe stress), followed by a failure to return to baseline.
That pattern of “perturbation followed by failure to reset” is exactly what a setpoint shift looks like. The chronicity, the resistance to intervention, and the hallmark symptom of post-exertional malaise (where even small perturbations push the system further into dysfunction rather than triggering recovery) all look like a system operating around a new, pathological attractor state.
In normal physiology, exercise triggers transient inflammation, followed by recovery, followed by adaptation to a stronger baseline. In ME/CFS, exercise triggers an exaggerated transcriptional immune response, followed by a delayed crash, followed by prolonged recovery. The system is not broken in the sense that a part is missing. It is stuck, locked into a defensive operating mode that it cannot exit.
How might the setpoint change happen?
The following is purely speculative, but I think it represents nonetheless an explanation why the setpoint switch happens in some people but not others.
ME/CFS usually begins after a viral infection. The most documented of these is EBV. In fact, after mononucleosis, about 10% of infected people meet CFS criteria after 6 months, particularly if the infection happens in adulthood (vs. in childhood). Similarly, after the first COVID infection, about 10% (studies range from 5-20%) of people meet CFS criteria after 6 months. Other viruses for which this is reported are human herpes virus 6, CMV (another herpes virus), parvovirus B19, Ebola virus, and some enteroviruses.
What all of these viruses have in common is that they are slowly cleared by the body. Mononucleosis sometimes lasts for many weeks to months. Similarly, on the first COVID infection, people often test positive for weeks on end.
The more severe and longer the infection the higher the likelihood of developing a CFS-like syndrome.
In other words, the longer the cytokinemia (high cytokine levels) the higher the likelihood that the brain is “taught” that there is a dangerous environment and it enters “sickness mode” from which it is unable to exit. In a way, the prolonged cytokinemia coupled with the viral infection somehow “imprints” into the central nervous system (and perhaps even other systems).
This explains why CFS is very uncommon after influenza. While influenza is severe, the active illness duration is usually short and the body clears the virus relatively quickly, in contrast to, e.g., EBV and SARS-CoV-2.
After my first COVID infection four years ago, my CT value was positive/measurable for over 3 weeks. I was tired for about 1-2 months after, perhaps a very light version of LongCOVID, which I believe most people had (as most people cannot not tell whether they have 10-20% less energy for a couple of months).
In fact, even when flu symptoms feel intense (e.g., high fever), the median febrile phase is 3–5 days, viremia is minimal, and the immune system usually clears the virus quickly.
By contrast, EBV has a prolonged, systemic, lymphoproliferative phase (weeks), and SARS-CoV-2 can also persist for weeks in tissues and is associated with delayed viral clearance in some.
Duration of high cytokine activity seems to matter more than peak intensity. ME/CFS is strongly linked to aberrant immune reprogramming (chronic low-grade activation, poor NK cell function), which influenza rarely provokes. Furthermore, influenza mostly affects the respiratory epithelium whereas EBV and SARS-CoV-2 affect many other tissues (including neurons).
Individuals with CFS often but not always have a similar phenotype. They have a pronounced fatigue, particularly after exertion (often lasting for days and weeks), also known as “crashes”, they often have a high heart rate, postural hypotension (dysautonomia). Psychiatric symptoms secondary to the development of the disease are common (e.g., a tendency to be anxious). There also seems to be a host of other organ-specific symptoms (e.g., lung-specific symptoms in COVID-19, which though may have nothing to do with ME/CFS and might just represent damage from the disease independent of the setpoint reprogramming).
Judged from the basis of this hypothesis, it is also conceivable why ME/CFS is more likely to develop if one has too much physical exertion during the recovery phase. If people exercise too early, many long-COVID researchers agree that ME/CFS rates seem to be much higher.
Being depressed or anxious at baseline (before contracting ME/CFS) is a risk factor for the disease, possibly because depression and anxiety are linked to higher levels of neuroinflammation and/or reduced levels of neuroplasticity, which presumably make it easier for ME/CFS to take hold as the brain may be more vulnerable to “danger signals”.
On Reddit, there are countless anecdotes of people who overexerted themselves too early and are now stuck with CFS. Given the crappy state of ME/CFS research, anecdotes are not nothing. This could even be explainable physiologically – e.g., exercise leading to increased IL-6 levels or ATP-depletion, which then would tell the brain “it is getting worse, we need to enter/intensify sickness mode”.
It seems that with every crash, individuals with ME/CFS tend to get a little worse. It could be because exercise leads to a depletion in ATP and/or a spike in cytokine levels, both of which are interpreted as a “danger” signal telling the brain that it should better stay in “shutdown” mode.
Evolutionary origins
The body has deeply conserved programs for surviving existential threats: particularly starvation (leading to long-lasting changes in neuroendocrine energy systems), prolonged overwhelming stress (leading to long-lasting changes in vitality– aka depression) or severe infection (leading to long-lasting “sickness mode” – aka ME/CFS).
In each of these, a coordinated program is turned on to protect the organism.
In the case of starvation, it protects the organism against another famine (often seen in people with a past eating disorder). I discuss this in-depth in my article on The Sickness of the Ultra Fit.
In the case of depression, some researchers believe that biological depression may have evolved as a protective mechanism to prevent an organism from running itself into the ground. According to this theory, prolonged stress (or a prolonged effort-reward imbalance) triggers biological depression (a downward deviation from baseline) to “force” individuals emotionally to let go of unattainable goals and/or withdraw from desperate situations. If an individual’s energy and mood deteriorates she is more likely to let go and withdraw. Conversely, if an individual’s energy and mood did not deteriorate, she might continue to overexert herself and die. Indeed, prolonged stress (i.e., perceived effort) seems to be one of the greatest risk factors for developing depression. I discuss this in more detail here: Depression – Basic Principles.

In the case of ME/CFS, a “sickness mode” program is turned on and then fails to turn off again. This program involve coordinated downregulation of energy expenditure, suppression of non-essential functions, heightened immune surveillance, and metabolic conservation. They are adaptive in the short term. The problem in ME/CFS may be that these programs fails to turn off.
Robert Naviaux’s metabolomics work (linked below) has shown that ME/CFS patients exhibit a hypometabolic signature resembling what he calls the “cell danger response,” essentially a conserved cellular state analogous to the state that certain organisms enter under threat. If mitochondrial function and cellular energy metabolism can be epigenetically downregulated across multiple tissues, that would look like a shifted metabolic setpoint, and it maps directly onto the core symptom of profound energy limitation.
This framing unifies several otherwise disparate findings. The immune abnormalities in ME/CFS (altered cytokine patterns, reduced NK cell function, autoantibodies to adrenergic and muscarinic receptors, a mild chronic inflammatory signature) are real, but they are not extreme, and they are inconsistent across patient cohorts. They do not fully explain the central phenomenon of post-exertional malaise. If you think of immune dysregulation as the trigger rather than the engine, this makes sense.
The infection (or other initial insult) provides the spark, the massive inflammatory stress that pushes the system past a tipping point. But the engine, the thing that keeps the disease going, is the new setpoint itself: a self-sustaining configuration of gene expression, autonomic tone, and neuroimmune signaling that the body now defends as its new normal.
“Where” is the setpoint shifted?
If this model is correct, the most likely location for the critical epigenetic changes is the central nervous system, which is, unfortunately, largely inaccessible in living patients. Several candidates stand out.
Microglial priming is one strong possibility. Microglia (the brain’s resident immune cells) can undergo epigenetic reprogramming after an initial activation event, a phenomenon sometimes called “trained immunity”. This leaves them in a sensitized state where they overreact to subsequent stimuli. A persistently primed microglial population could maintain low-grade neuroinflammation that disrupts circuit function throughout the brain without producing the kind of overt pathology visible on standard imaging. This has been demonstrated in rodent models of post-infectious neuroinflammation, and it would explain the brainstem abnormalities and altered microglial activation that have been observed in ME/CFS patients.
Autonomic set-point shifts are another candidate. The autonomic nervous system controls heart rate, blood pressure, digestion, and energy allocation, and autonomic dysfunction (POTS, orthostatic intolerance) is one of the most consistent features of ME/CFS. If the regulatory circuits governing autonomic tone have been epigenetically reprogrammed to a new operating point, this would explain why the dysfunction is so pervasive and so resistant to treatment.
There is also precedent in related conditions. Central sensitization in chronic pain states (including fibromyalgia, which shares many features with ME/CFS) involves persistent changes in how the nervous system processes sensory signals, and epigenetic mechanisms have been implicated. PTSD involves a persistent reset of threat-detection thresholds. These are all examples of the same fundamental phenomenon: a biological system that has been pushed into a new stable state and lacks the mechanism to return.
One more connection worth mentioning. Post-finasteride syndrome (a condition where men develop persistent sexual, neurological, and cognitive symptoms after discontinuing the hair loss drug finasteride) may share the same fundamental mechanism: a persistent regulatory state change after a pharmacologic perturbation. Finasteride alters steroid hormone metabolism, and in susceptible individuals, the regulatory system appears to fail to revert after the drug is withdrawn. The phenomenology (persistent symptoms, resistance to treatment, a “stuck” quality) is strikingly similar to ME/CFS. Both could represent maladaptive regulatory reprogramming, the same kind of setpoint shift triggered by different initial insults. Anecdotally, the way to reverse PFS seems to be to “bombard” the brain with androgenic steroids (particularly DHT-derivatives), which may seem to be able to revert “stuck” transcriptional loops.
So, what are the androgenic steroids of ME/CFS? Prolonged use of potent anti-inflammatory drugs (e.g., a combination of monoclonal antibodies targeting individual cytokines)?
Reframing the therapeutic question
If ME/CFS is a defended setpoint, then the therapeutic question changes fundamentally. Instead of asking “what is broken and how do we fix it?” we should be asking “what is maintaining the new operating point, and how do we destabilize it?”
This explains why so many promising interventions have produced disappointing results. Purely suppressing inflammation may not work because the inflammation is a downstream consequence of the setpoint, not its cause. Purely boosting mitochondrial function may not work because the mitochondrial suppression is being actively maintained by regulatory signals (i.e., self-maintaining feedback loops of transcription factors). Purely treating autoimmunity may not work because the autoimmune features are epiphenomena of a deeper dysregulation.
The analogy to obesity is instructive here too. We do have one intervention that can shift the obesity setpoint (at least partially): bariatric surgery. Some patients achieve sustained, dramatic weight loss after surgery, which suggests the defended weight can be moved downward. If the mechanism were truly irreversible (e.g., complete neuron death), this would be impossible. The fact that it works points toward functional impairment (most likely candidate: epigenetic changes in key neuronal populations) rather than permanent structural destruction of something.
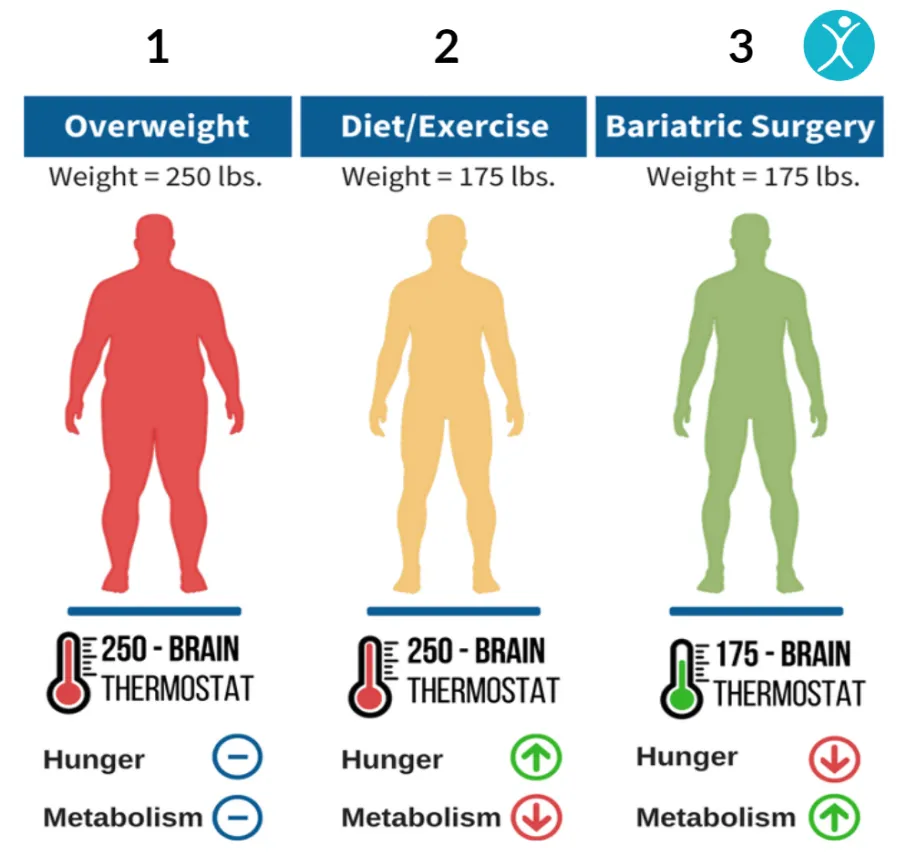
So the question becomes: what is the “bariatric surgery” of ME/CFS? What intervention could be dramatic enough to destabilize the pathological setpoint and allow the system to reorganize around a healthier operating point?
A possible path forward: precision epigenetic editing
I believe the most promising long-term approach (and I acknowledge this is still largely future technology) is precision epigenetic editing. In research settings, scientists are already using tools like dCas9 (a version of the CRISPR-associated protein that binds DNA without cutting it) fused to methyltransferases or histone acetyltransferases, as well as targeted chromatin remodeling enzymes. These tools allow specific genes to be turned up or down without altering the DNA sequence itself.
This is, theoretically, exactly what you would need to reset a microglial inflammatory program, silence a chronic interferon signature, or break a metabolic trap gene circuit. If we could identify the specific epigenetic changes maintaining the ME/CFS setpoint (which genes are hypermethylated or hypomethylated, which histone marks are altered, in which cell types), we could, in principle, reverse them directly.
We are not there yet. The technology needs to mature, we need better tools for delivering epigenetic editors to specific cell populations in the CNS, and (most critically) we need to identify the precise epigenetic targets. But the framework suggests where to look and what to look for, which is more than we had before.
The uncomfortable possibility
I should mention a darker possibility that I think is less likely but cannot be ruled out. Could the prolonged cytokinemia and neuroinflammation caused by certain viruses (EBV, SARS-CoV-2) actually cause the death of specific “central” neurons involved in energy regulation, autonomic control, or neuroimmune coordination?
If so, ME/CFS would be more analogous to a neurodegenerative condition, and the setpoint shift would not be a functional lock but a structural loss. The brain might find ways to navigate around the damage (as it often does after a stroke), but dead neurons cannot undie. Sure, the brain does retain some potential of neurogenesis after destruction and it also certainly retains a lot of neuroplasticity, which is why some people may get better.
I personally find this possibility less likely for several reasons. ME/CFS patients do show fluctuations in severity, and some patients do recover (particularly in the early years), which is hard to reconcile with irreversible neuron loss. The condition also lacks the progressive worsening trajectory typical of neurodegenerative diseases. But it is a possibility that the field needs to take seriously, particularly for patients with very long disease duration.
Conclusion
The setpoint model does not solve ME/CFS. But it does something important: it provides a coherent framework that unifies the disparate findings in the field and reframes the therapeutic question in a way that might finally lead somewhere.
For too long, ME/CFS research has been fragmented, chasing individual abnormalities (immune markers, metabolic deficits, autonomic dysfunction, gut microbiome changes) without a unifying theory of what holds the disease together. The setpoint hypothesis suggests that these are all downstream manifestations of a single deeper phenomenon: an epigenetically locked neuroimmune operating point that the body defends as its new normal.
The analogy to obesity is not perfect, but the underlying logic (epigenetic locking of homeostatic circuits into pathological operating points) is biologically plausible and could unify a lot of the disparate findings in the field. And it suggests that the path forward lies not in treating individual symptoms or downstream abnormalities, but in understanding and ultimately destabilizing the defended state itself.
As long as the field keeps looking in directions that address symptoms rather than the core mechanism, we will continue to get the low-quality answers we got thus far.
I want to emphasize again that this is only a hypothesis, but nonetheless a hypothesis worth exploring.
Sources and further information
- Naviaux RK et al. “Metabolic features of chronic fatigue syndrome.” PNAS 113(37):E5472–E5480 (2016).
- Naviaux RK. “Metabolic features of the cell danger response.” Mitochondrion 16:7–17 (2014).
- Naviaux RK. “Metabolic features and regulation of the healing cycle — A new model for chronic disease pathogenesis and treatment.” Mitochondrion (2018).
- Loebel M et al. “Antibodies to β adrenergic and muscarinic cholinergic receptors in patients with Chronic Fatigue Syndrome.” Brain Behav Immun 52:32–39 (2016).
- Bynke A et al. “Autoantibodies to beta-adrenergic and muscarinic cholinergic receptors in Myalgic Encephalomyelitis (ME) patients — A validation study.” Brain Behav Immun Health 7:100107 (2020).
- Scheibenbogen C et al. “Efficacy of repeated immunoadsorption in patients with post-COVID ME/CFS and elevated β2-adrenergic receptor autoantibodies.” Lancet Regional Health – Europe (2024).
- Plagemann A et al. “Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: an epigenetic model of obesity and the metabolic syndrome.” J Physiol 587(Pt 20):4963–4976 (2009).
- Ramamoorthy TG et al. “Maternal overnutrition programs epigenetic changes in the regulatory regions of hypothalamic Pomc in the offspring of rats.” Int J Obesity 42:1431–1444 (2018).
- Kühnen P et al. “An Alu element-associated hypermethylation variant of the POMC gene is associated with childhood obesity.” PLoS Genetics 8:e1002543 (2012).
- Lechner L et al. “Early-set POMC methylation variability is accompanied by increased risk for obesity and is addressable by MC4R agonist treatment.” Science Translational Medicine (2023).
- Kühnen P et al. “Epigenetic regulation of POMC; implications for nutritional programming, obesity and metabolic disease.” Mol Cell Endocrinol (2019).
- Wendeln AC et al. “Innate immune memory in the brain shapes neurological disease hallmarks.” Nature 556:332–338 (2018).
- Neher JJ, Cunningham C. “Priming microglia for innate immune memory in the brain.” Trends Immunol 40:358–374 (2019).
- Martins-Ferreira R et al. “Microglial innate memory and epigenetic reprogramming in neurological disorders.” Prog Neurobiol 200:101971 (2021).
- Ayambem Nana D et al. “Microglial immune regulation by epigenetic reprogramming through histone H3K27 acetylation in neuroinflammation.” Front Immunol 14:1052925 (2023).
- Holtman IR et al. “Epigenetic regulation of innate immune memory in microglia.” J Neuroinflammation 19:111 (2022).
- Denk F, McMahon SB. “Chronic pain: emerging evidence for the involvement of epigenetics.” Neuron 73(3):435–444 (2012).
- Global Burden of Disease Long COVID Collaborators. “Estimated global proportions of individuals with persistent fatigue, cognitive, and respiratory symptom clusters following symptomatic COVID-19 in 2020 and 2021.” JAMA 328(16):1604–1615 (2022).
Disclaimer
The content available on this website is based on the author’s individual research, opinions, and personal experiences. It is intended solely for informational and entertainment purposes and does not constitute medical advice. The author does not endorse the use of supplements, pharmaceutical drugs, or hormones without the direct oversight of a qualified physician. People should never disregard professional medical advice or delay in seeking it because of something they have read on the internet.