
Currently, about 50% of all deaths in industrialized nations are due to atherosclerosis, which is also the condition that kills most humans worldwide.
Preventing atherosclerosis is crucial for longevity
A lot of solving the longevity equation comes down to delaying the onset of atherosclerosis. The earlier risk factors are addressed, the better the chance to delay (or ideally, prevent) this disease and its consequences later in life. The consequences of atherosclerosis go far beyond cardiovascular events (such as heart attack, and stroke) and include a worsening blood supply to essentially all cells of the body.
The most well-known form of this is peripheral artery disease, which includes pain while walking (intermittent claudication) and poorly healing infections often leading to amputation. However, atherosclerosis progressively worsens blood supply even if the patient does not notice – which is often overlooked. The first signs are sometimes erectile dysfunction (worsening of blood supply to the penis) and even dementia (worsening of the blood supply to the brain), however, ultimately, blood supply is worsened to every cell, tissue, and organ because atherosclerosis is a systemic condition of the arterial system.
Unless humanity is able to fundamentally change how lipoproteins interface with the endothelium, this disease will progress in everyone, although the rate of progress can be vastly modified. The earlier the intervention, the better, as the longer my artery walls are exposed to atherogenic particles and other risk factors, the more likely they are to incur damage.
How atherosclerosis develops
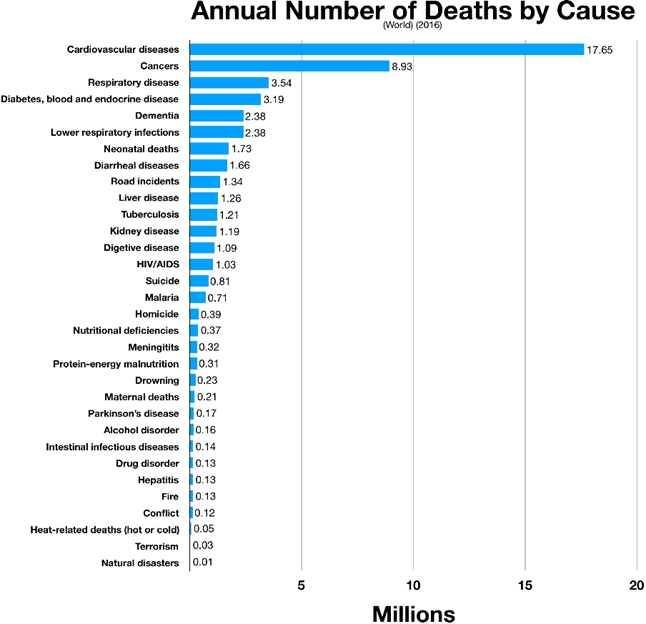
Atherosclerosis (“cardiovascular disease”) is a time-course disease, which is why age is such a strong predictor of risk. The longer arterial walls are exposed to insults and atherogenic particles (lipoproteins) the more damage they will incur.
Atherosclerosis is fundamentally a progressive narrowing and degeneration of blood vessels. This disease starts already in the first decade of life and eventually kills people via heart attack, heart failure, or stroke.
There are multiple drivers that can lead to the development of atherosclerosis. The most widely known are high blood lipids (dyslipidemia), high blood pressure (hypertension), high blood sugar (hyperglycemia), and smoking. These so-called “Framingham factors” factors (excluding smoking) are often a direct consequence of metabolic disease.
Some of the contributing factors are high levels of Lp(a), low-level inflammation, hypercortisolemia, or low levels of thyroid hormones, sex hormones, or growth hormone/IGF-1. Furthermore, there are a large variety of genes that are thought to influence atherogenesis in one way or another. In fact, genetics presumably play a huge role. Some people live well into their 90s with sky-high levels of ApoB or lp(a) whereas others have CV-events early, without any obvious risk factors.
As always, biology is messy: Multiple things occur in parallel, and everyone has a unique constellation of protective and predisposing factors.

How does atherosclerosis happen?
The first step in atherosclerosis is injury to the innermost lining of blood vessels (endothelium) by a variety of mechanisms, including smoking, hypertension, or hyperglycemia. The ensuing inflammatory reaction then inflames and thickens the endothelium (“intima thickening”).
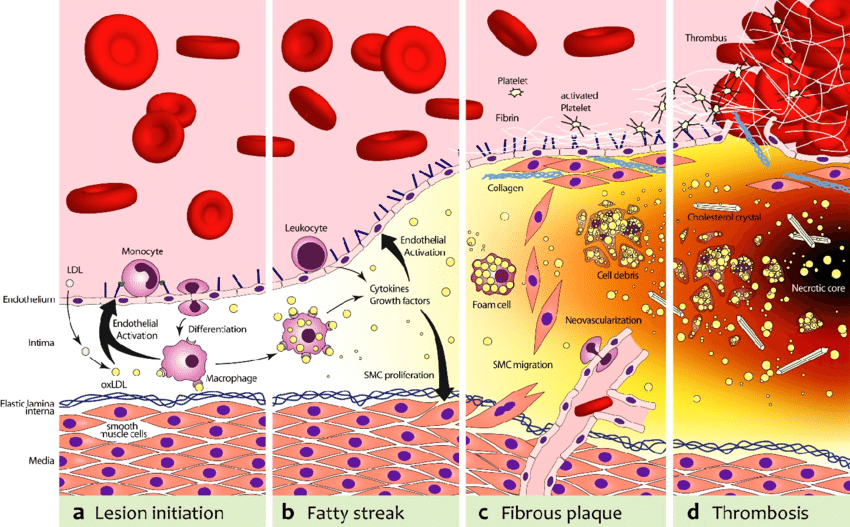
At the same time, lipoproteins (essentially LDL particles) get into the inflamed vascular wall. If these LDL particles come in contact with the pre-damaged vascular wall, they are oxidized. (Technically, LDL particles by themselves are thought to be harmless, and the ultimate atherogenic compounds are oxidized molecules contained within them.) These oxidized LDL particles are then engulfed by macrophages. Macrophages die and cause even more inflammation. Fibrosis (scar tissue) develops and narrows the artery.
In addition to the narrowing, depending on how bad the inflammatory environment is, a lipid-laden cap starts to cover the lesion (“plaque”), which renders it vulnerable to rupture.
Upon rupture, blood clotting can occlude the artery and cause an infarction (e.g., if a cardiac artery is occluded this is called a heart attack). Alternatively, the ruptured lipid-laden mess can be carried away and occlude an artery somewhere downstream (e.g., a plaque that forms in the heart atria dislodges and is transported into the circulatory system of the brain causing a stroke.)
Risk factors
Unfortunately, there are many risk factors at play:
- A large number of genes
- The number of lipoproteins cruising through the bloodstream at any given time (measured by ApoB – the protein found on the surface of atherogenic lipoproteins mostly responsible for driving atherosclerosis)
- Lp(a) particles (LDL particles with an apolipoprotein(a) attached to them)
- Anything that causes injury to the endothelium. First and foremost, high blood pressure, high blood sugar, and smoking.
- A variety of hormones, all of which have a variety of direct and indirect effects on endothelial health, metabolic health, and repair capabilities.
- Levels of inflammation
There are many other “obscure” genetic factors at play that are poorly understood
Atherosclerosis can run in families even when there’s no obvious cause. They don’t have FH (familial hypercholesterolemia) or high Lp(a). Their apoB is around the 50th percentile of the population. Yet, they are disproportionately afflicted at a young age – meaning, a heart attack in their fourties.
Similarly, there are tons of patients who’ve made it to their 90s with high apoB levels (and even high lp(a) levels) that don’t have obvious atherosclerotic disease. Nonetheless, the evidence is quite clear and we understand atherosclerosis better than most other multifactorial diseases in medicine. Just because some people can get away with chain-smoking for life does not mean that chain-smoking is not the root cause of most cases of COPD or lung cancer.
Tactics I follow to delay atherosclerosis
- Blood pressure
- Managing lipoproteins
- Hormone optimization
- Metabolic health
- Avoiding spikes in blood sugar
- Keeping insulin levels fairly low
- Maintaining a low body fat percentage
- Optimizing endothelial health
- Lowering inflammation
- Exercise
- Lp(a)
- TMAO & homocysteine
Keeping an eye on blood pressure
Out of all the Framingham factors, hypertension is the deadliest. It is thought that hypertension kills about 9 Mio. people per year, which is perhaps 10-15% of all deaths. For every 10mmHg reduction in RR, the risk for cardiovascular events drops by 45% (data derived from Mendelian randomization).
Blood pressure leads to atherosclerosis through injury to the intima, the innermost lining of arteries. This then leads to intima thickening and atherosclerotic progression (as explained above).
High blood pressure runs in my family and I have a couple of (potentially) deleterious single nucleotide polymorphisms (SNPs) in my renin-angiotensin-aldosterone system (RAAS). However, fortunately, my blood pressure is usually around 120/65 mmHg.
Obviously, weight control and exercise, both of which have independent effects on blood pressure, should be taken care first. However, blood pressure has strong genetic underpinnings.
If I were to choose a drug for hypertension, I would choose telmisartan over other antihypertensives. Telmisartan is an angiotensin receptor blocker (ARB). It has a long half-life and beneficial effects on metabolic health because of its off-target effects on PPAR-delta (a transcription factor involved in fatty acid metabolism). PPAR-delta modulators are banned by WADA because of their effects on endurance capacity. Unfortunately, most doctors choose ramipril or valsartan as their first-line antihypertensive – mostly because of historical reasons.
Of note, automatic RR-measuring devices often overestimate the systolic pressure by 5-10mmHg.
Managing lipoproteins
The lower lipoproteins are (particularly LDL levels) the better. Kids have very low LDL levels (roughly 20-30mg/dl) and they do not suffer any adverse effects. Similar things hold true for people with a mutation in the PCSK9 gene (hypofunction). They have very low LDL levels (sometimes below 20mg/dl) but do not have any increased risk of dementia, depression, or any other disease – but they are virtually free of atherosclerosis (including heart attacks and many forms of stroke).
As explained above, lipoproteins (essentially LDL particles) get into the inflamed vascular wall where they are oxidized. These oxidized LDL particles are then engulfed by macrophages. Macrophages die and cause even more inflammation. Fibrosis (scar tissue) develops and narrows the artery, often with a lipid-laden cap.
Consequently, lipoprotein particles are the causative agent of atherosclerosis (“necessary but not sufficient”). For every 40mg/dl reduction in LDL, the risk of CV events drops by 55% (Mendelian randomization data). Similarly, a reduction of 25mg/dl LDL + a reduction of 5mmHg RR reduces the risk by 55% as well.
Atherogenesis is sterol-mediated, but sterols are trafficked within ApoB-containing lipoproteins. In other words, it is the number of ApoB particles that is most relevant. Hence, ApoB is the blood marker one should test for.
Since I started doing regular blood tests about a decade ago, my ApoB levels are consistently below the clinical reference range (ApoB between 35-50mg/dL and my HDL at around 70mg/dL – though it is becoming clear that HDL is a mostly worthless marker). Interestingly, no matter what diet I follow (e.g., keto, intermittent fasting, high-carb diet) my ApoB levels do not seem to change much.
This means that I am naturally in the top 2.5% of the population. Initially, I thought they were that low because of all the drugs and supplements I experiment with but I eventually found out that my brother, who barely takes anything, also has ultra-low levels. So I guess I am blessed in this regard (I discuss my genetic test results in more detail here). However, fish oil and rapamycin may drive them even lower.
If my ApoB levels were an issue, I would not even bother with lifestyle interventions (which do not do much for lipids anyway) but would immediately jump to 2.5mg of rosuvastatin (half of the lowest clinical dose) plus 10mg of ezetimibe.
Rosuvastatin is a statin (blocking HMG-CoA-Reductase – one of the key enzymes in the synthesis of cholesterol) and at low doses, it is mostly liver-targeted. Furthermore, it is very hydrophilic compared to other statins and the lack of lipophilicity keeps it mostly outside the blood-brain barrier.
2.5mg of rosuvastatin is known to knock down ApoB levels by about 40%. 40mg of the drug only achieves about 55% despite being 16x the dosage (consequently with more systemic effects). Unfortunately, in the clinic, I see 40mg of rosuvastatin given out all the time.
A friend who is statin-intolerant has capsules containing 1mg of rosuvastatin compounded for himself, which he can take without side effects (for reference: the lowest clinical dose is usually 5mg).
In my opinion, from an angle of primary prevention, there is little need for clinical doses, which come with a greater incidence of adverse effects but do not lower LDL by much more anyway. Statins are also thought to lower inflammation, as evidenced by a decrease in CRP levels upon starting statin treatment.
Three other drugs commonly used for lowering LDL particle count are:
- Ezetimibe – a blocker of the absorption of endogenous cholesterol. Can be added to all the other lipid-lowering agents. Mostly devoid of side effects.
- For statin-intolerant people there is bempedoic acid. An amazing drug, though unfortunately quite expensive and/or hard to get approved for.
- Then there are the mighty (but even more expensive) PCSK9 inhibitors, blockers of an enzyme responsible for the recycling of LDL receptors. They are also known to lower lp(a) levels by about 30%. Recently, inclisiran has been added to the arsenal. Inclisiran is a siRNA that needs to be injected every six months. It also inhibits the synthesis of PCSK-9 but by a different mechanism than PCSK9 inhibitors.
There are benefits of starting lipid-lowering therapies and blood pressure-lowering therapies as early as possible as the progression of atherosclerosis is both a function of apoB concentration & blood pressure and duration of exposure. This is similar to smoking, where the damage is cumulative and measured in “pack-years” (1 pack-year being 1 year of smoking one pack per day).
Of note, HDL (also known as “good” cholesterol) is no longer considered informative to use and a higher HDL is associated with better lipd levels but it may not have any protective effect by itself.
Hormone optimization
Thyroid hormones, growth hormone/IGF-1, and sex hormones are all quite highly anti-atherogenic. I make sure that all of these are “good” (youthful levels). Once my other hormones are going to decline, I will seriously consider replacing them. Hormones are discussed here.
For women, atherosclerosis vastly accelerates after menopause. Before menopause, E2 acts as a potent protective factor. On average, women still have a delay in atherosclerosis by about 10y.
Metabolic health
Metabolic health is discussed in depth in another article. In short, my strategies include keeping body fat levels low, endurance exercise, HIIT, maintaining a decent amount of muscle mass, rapamycin, optimizing hormones, keeping insulin sensitivity high year-round, diet, sleep, and metabolic drugs.
Avoiding spikes in blood glucose
I try my best to avoid spikes in blood glucose whenever I am eating by myself – I do not care about spiking blood sugar during social occasions.
Whenever levels of blood glucose are high, glucose covalently bonds to certain amino acids on proteins (Maillard reaction) leading to the formation of advanced glycogen end products (AGEs). These AGEs are phagocytized by macrophages through the RAGE-receptor (among others), which stimulates inflammation.
Furthermore, due to the covalent crosslinking, protein structure (and therefore function) is compromised. Both of these (AGEs; protein function) play a prominent role in micro-vascular disease, which is among the leading causes of blindness, renal failure, and neurodegeneration. Furthermore, it accelerates macrovascular disease (atherosclerosis).
Keeping insulin levels fairly low
While insulin will lower blood glucose (which is beneficial for micro-vascular disease), insulin itself promotes the development of macro-vascular disease (atherosclerosis), which includes coronary vascular disease (leading to heart attack), cerebrovascular disease (leading to stroke), and peripheral vascular disease (leading to amputation).
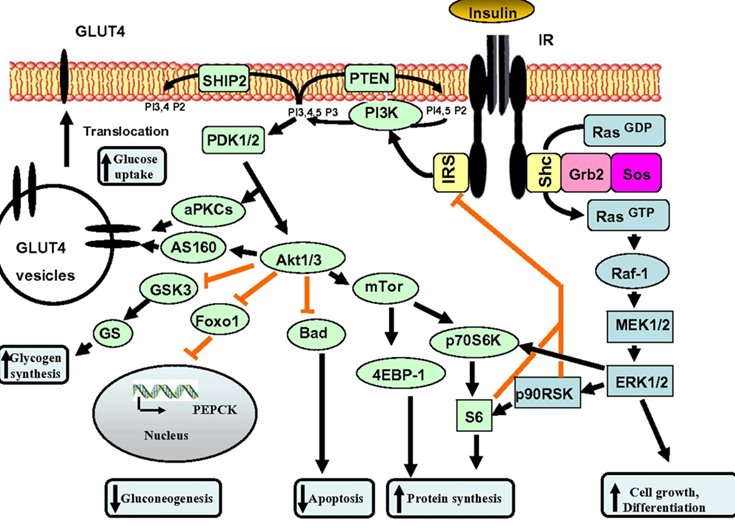
Unfortunately, few physicians realize that controlling blood glucose (HBA1C) is only half of the job, and the other half is about keeping area-under-curve levels of insulin low. A lot of this comes down to insulin being a potent growth factor, and “insulin all of the time” equals “growth all of the time”, which is driving pretty much every chronic disease, including atherosclerosis.
The two best ways to keep area-under-curve insulin levels low without having to fast is to reduce consumption of fast-spiking carbohydrates (“bad carbs” – I usually only consume “bad carbs” after exercise, a time when my liver and muscle are primed to mop up excess glucose to resynthesize glycogen) and to keep insulin sensitivity high. I discuss the strategies I use to keep insulin sensitivity high here.
Maintaining low body fat percentage
I try to keep my body fat low. This keeps me insulin-sensitive and lowers lipid levels in my bloodstream because adipocytes are primed to absorb them. A better lipid profile (essentially lower levels of ApoB and triglycerides) means less atherosclerosis.
Things that help me with this are a hefty dose of exercise, adhering to a couple of dietary principles, optimizing my hormones, and a very low dose metreleptin and on-and-off use of semaglutide .
Optimizing endothelial health
Similar to how it is hard to define what “metabolic health” is, it is hard to define what endothelial health is. I try to keep my endothelium healthy by optimizing hormones, rapamycin, my past use of senolytics (?), antioxidants, and exercise.
I experimented on and off with a very low dose of tadalafil (1mg/d) to increase cardiovascular health and for its putative anti-inflammatory benefits. Tadalafil is a PDE5 inhibitor, frequently used for erectile dysfunction.
The idea is that the boost in nitric oxide signaling will deliver a lot of cardiovascular benefits, which is also presumably one reason why eating vegetables is associated with better cardiovascular health (vegetables contain a lot of nitrates, some of which convert into nitric oxide).
Furthermore, PDE5 inhibitors have also been shown to have anti-inflammatory, antioxidant, antiproliferative, and metabolic-health-enhancing properties in several experiments.
However, while they likely did improve my endothelial health (at least as judged by my forearm vascularity as a pathetic proxy), my most recent experiment led to a sudden onset of tinnitus within 30min of taking 1mg of tadalafil.
Given that PDE5 inhibitors are highly associated with new-onset tinnitus and weakly associated with hearing damage, I decided to stop tadalafil for good, as even a small chance of sensory impairment (and therefore loss of quality of life) is not worth a small cardiovascular benefit in an otherwise healthy young person. Fortunately, the tinnitus went away after 48 hours.
“Fun” fact: Hugh Hefner, the founder of Playboy, blames his almost complete hearing loss on the overuse of sildenafil (Viagra), and there is reason to believe that there is a causal correlation.
Lp(a)
As I was working in cardiac and vascular surgery, there were lots and lots of amputations happening all the time. When patients are lucky, it is just a toe. But most of the time, it is the whole foot or lower leg.
Most of the patients have a combination of long-standing poorly controlled hyperglycemia and full-time smoking. The endothelial inflammation and the neuropathy lead to non-recognized and poorly healing infections which may eventually turn into sepsis – hence the preemptive amputation.
One time, however, there was a lower leg amputation on a guy who is barely 45 years old. He was not even overweight and did not have diabetes. While he was a smoker, the primary reason for losing his lower leg at such a young age was elevated levels of lp(a).
In his case, the elevated levels of lp(a) led to early atherosclerotic degeneration of blood vessels, including manifest peripheral artery disease. A small injury led to a poorly healing infection, which turned into the requirement for amputation.
Lp(a) is an insidious particle that is much more important than commonly appreciated. Through pro-inflammatory mechanisms, it drives the development of atherosclerosis. Patients with high levels of lp(a) often have major cardiovascular events (e.g., heart attack, stroke) and peripheral artery disease at a young age.
About 10% of people have genetically elevated Lp(a), which may have conferred an advantage in blood clothing in the past. Again, evolution favored something that is awesome for the young at the expense of the old. There is quite little one can do to lower lp(a) beyond turning to cutting-edge pharmacology.
Fortunately, my Lp(a) is naturally low (around 10mg/dl). If my Lp(a) were high, I would turn to PCSK9 inhibitors to lower it. There is also an antisense drug on the way that will hopefully be available for clinical use soon (pelacarsen). There are also various gene therapies currently in various phases of clinical development.
TMAO & homocysteine
Next to lp(a), also TMAO & homocysteine are associated with atherosclerotic risk.
TMAO is a small amine molecule found in many marine species to help preserve protein stability at great depths. It is thought that in mammals, TMAO activates macrophages, thereby leading to inflammation, which then promotes atherogenesis but the exact mechanism is still unclear.
In humans, TMAO can be produced by certain gut microbes in response to carnitine and cholines (e.g., alpha-GPC, phosphatidylcholine). I supplement with both of them. Unfortunately, thus far, I have not been able to find a laboratory that measures TMAO levels.
Homocysteine is an amino acid produced as a byproduct during the metabolism of methionine (an essential amino acid). It is thought that homocysteine is an autoreactive molecule that produces oxidative stress. High levels of homocysteine seem to be correlated with cardiovascular disease. Fortunately, my homocysteine levels always came back low when I tested for them. TMG, a supplement I take, is known to lower homocysteine levels as are methylated B vitamins.
However, in comparison to the importance of blood pressure & ApoB & lp(a), TMAO & homocysteine are barely worth mentioning!
Lowering inflammation
Discussed extensively here.
Exercise
I do about 1 hour of exercise per day. Usually, I alternate between the gym and steady-state cardio (so-called “zone II exercise”). Twice per week I also work on my mobility. I discuss my exercise regimen in more detail here.
My Longevity Protocol (Long & Technical Version)
This article is part of a much larger post describing my complete longevity blueprint. For my full protocol, read here.
Longevity Protocols
- My General Longevity Framework
- My Most Likely Cause of Death – My Protocol for Fighting Atherosclerosis
- The Ultimate Frontier – My Protocol for Fighting Cancer
- My Protocol for Fighting Dementia – What I Do To Keep Brain Health Optimal
- My Approach to Hormone Optimization for Longevity
- Crucially Important & Underrated – My Protocol for Optimizing Metabolic Health
- What Is All the Fuss About mTOR?! – Why (and How) I Reduce the Activity of the mTOR Pathway
- Eliminating Zombie Cells – My Protocol for Fighting Senescent Cells
- Burning to Death – My Protocol for Lowering Inflammation
- My Physical Fitness Protocol
- My Protocol For Optimizing Organ Health
- Avoiding Toxins & Minimizing Infections
- My Protocol For Optimizing Mental Health
- What Genetic Tests Told Me About My Longevity
- Side Effects of My Longevity Protocol
Sources & further information
- Opinion article: Peter Attia – When does heart disease begin (and what this tells us about prevention)?
- Podcast: Peter Attia & Ron Krauss – A deep dive into heart disease
- Website: Ototoxic Adverse Drug Reactions: A Disproportionality Analysis Using the Italian Spontaneous Reporting Database
Disclaimer
The content on this website represents the opinion and personal experience of the author and does not constitute medical advice. The author does not endorse the use of supplements, pharmaceutical drugs, or hormones without a doctor’s supervision. The content presented is exclusively for informational and entertainment purposes. Never disregard professional medical advice or delay in seeking it because of something you have read on the internet.